Austin's paper (Wang et al. 2020 AJHG) on allele-specific fine mapping is out! 🎉
Austin’s paper on PLASMA, a new method for fine-mapping causal variants from molecular data using using allele-specific signals is now in print at The American Journal of Human Genetics. Congrats Austin et al!
Applied to RNA-seq data from 524 kidney tumor samples, PLASMA achieves a greater power at 50 samples than conventional QTL-based fine mapping at 500 samples, with more than 17% of loci fine mapped to within five causal variants, compared to 2% by QTL-based fine mapping.
Allele-Specific QTL Fine Mapping with PLASMA.
AT Wang, AH Shetty, E O’Connor, C Bell, MM Pomerantz, ML Freedman, A Gusev. The American Journal of Human Genetics. 2020
Abstract: Although quantitative trait locus (QTL) associations have been identified for many molecular traits such as gene expression, it remains challenging to distinguish the causal nucleotide from nearby variants. In addition to traditional QTLs by association, allele-specific (AS) QTLs are a powerful measure of cis-regulation that are concordant with traditional QTLs but typically less susceptible to technical/environmental noise. However, existing methods for estimating causal variant probabilities (i.e., fine mapping) cannot produce valid estimates from asQTL signals due to complexities in linkage disequilibrium (LD). We introduce PLASMA (Population Allele-Specific Mapping), a fine-mapping method that integrates QTL and asQTL information to improve accuracy. In simulations, PLASMA accurately prioritizes causal variants over a wide range of genetic architectures. Applied to RNA-seq data from 524 kidney tumor samples, PLASMA achieves a greater power at 50 samples than conventional QTL-based fine mapping at 500 samples, with more than 17% of loci fine mapped to within five causal variants, compared to 2% by QTL-based fine mapping, and a 6.9-fold overall reduction in median credible set size compared to QTL-based fine mapping when applied to H3K27AC ChIP-seq from just 28 prostate tumor/normal samples. Variants in the PLASMA credible sets for RNA-seq and ChIP-seq were enriched for open chromatin and chromatin looping, respectively, at a comparable or greater degree than credible variants from existing methods while containing far fewer markers. Our results demonstrate how integrating AS activity can substantially improve the detection of causal variants from existing molecular data.
The method & code is available in the PLASMA repository.
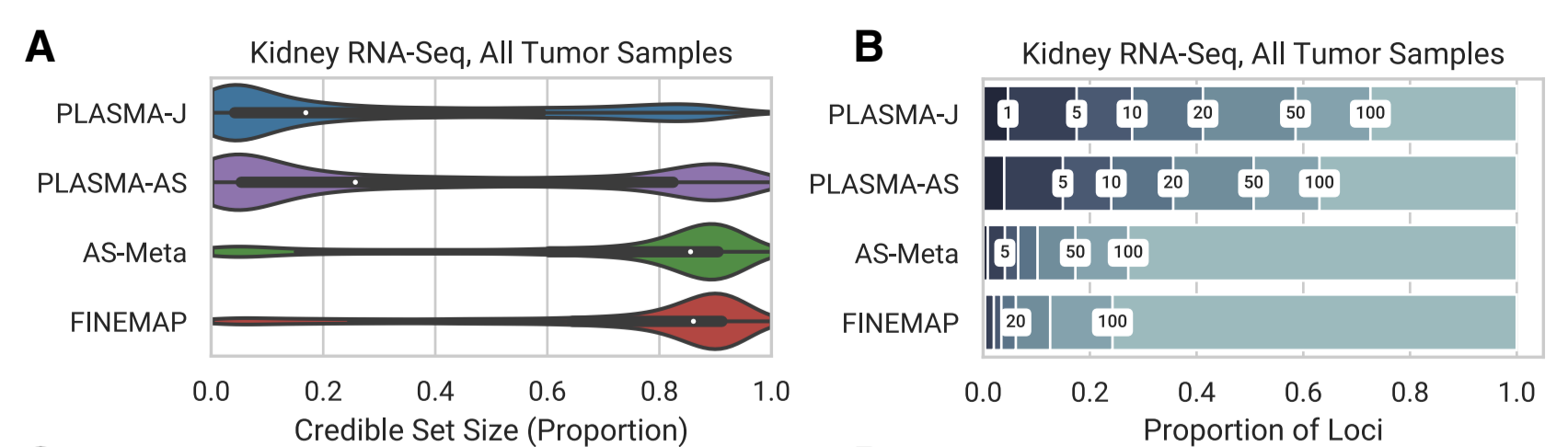
Comparison of the Distribution of 95% Confidence Credible Set Sizes across Loci in Kidney RNA-seq