New! RWAS (Grishin et al.) models for TCGA ATAC-seq
New! CONTENT (Thompson et al.) context-specific models for single-cell and bulk expression
New! GTEx v8 models
FUSION is a suite of tools for performing transcriptome-wide and regulome-wide association studies (TWAS and RWAS). FUSION builds predictive models of the genetic component of a functional/molecular phenotype and predicts and tests that component for association with disease using GWAS summary statistics. The goal is to identify associations between a GWAS phenotype and a functional phenotype that was only measured in reference data. We provide precomputed predictive models from multiple studies to facilitate this analysis.
Please cite the following manuscript for TWAS methods:
| Gusev et al. “Integrative approaches for large-scale transcriptome-wide association studies” 2016 Nature Genetics |
Please cite the following manuscript for RWAS methods and models:
| Grishin et al. “Allelic imbalance of chromatin accessibility in cancer identifies candidate causal risk variants and their mechanisms” 2022 Nature Genetics (accepted) |
For interactive analyses of hundreds of GWAS studies see www.twas-hub.org.
For questions or comments, contact Sasha Gusev [alexander_gusev@dfci.harvard.edu].
Download pre-computed predictive models
Compute your own predictive models
Joint/conditional tests and plots
wget https://github.com/gusevlab/fusion_twas/archive/master.zip
unzip master.zip
cd fusion_twas-master
wget https://data.broadinstitute.org/alkesgroup/FUSION/LDREF.tar.bz2
tar xjvf LDREF.tar.bz2
wget https://github.com/gabraham/plink2R/archive/master.zip
unzip master.zip
install.packages(c('optparse','RColorBrewer'))
install.packages('plink2R-master/plink2R/',repos=NULL)
if computing your own weights, the following additional steps are required
gcta_nr_robust to path (coded by Po-Ru Loh for robust non-linear optimization)plink to pathinstall.packages(c('glmnet','methods'))
ln -s ./ output in the directory where you will run FUSION.weights.R (this is a workaround because GEMMA requires results to go into an output subdirectory).The typical TWAS analysis takes pre-computed gene expression weights (below) together with disease GWAS summary statistics to estimate the association of each gene to disease. As an example, we will use the PGC Schizophrenia summary statistics to perform a TWAS with the GTEx whole-blood data. The example assumes you have setup FUSION and LD reference data as above and are in the FUSION directory with an LDREF subdirectory.
First, download and prepare the GWAS and GTEx whole blood data:
wget https://data.broadinstitute.org/alkesgroup/FUSION/SUM/PGC2.SCZ.sumstats
mkdir WEIGHTS
cd WEIGHTS
wget https://data.broadinstitute.org/alkesgroup/FUSION/WGT/GTEx.Whole_Blood.tar.bz2
tar xjf GTEx.Whole_Blood.tar.bz2
The WEIGHTS directory should contain a subdirectory of expression weights (which you can inspect in R), as well as several report files that describe the data (see below for details). The following sections describe the inputs in detail.
The primary input is genome-wide summary statistics in LD-score format. At minimum, this is a flat file with a header row containing the following fields:
SNP – SNP identifier (rsID)A1 – first allele (effect allele)A2 – second allele (other allele)Z – Z-scores, sign with respect to A1.and subsequent data rows for each SNP (all white-space separated). Additional columns are allowed and will be ignored. We recommend using the LDSC munge_stats.py utility for converting GWAS summary data into this format, which detects and reports many common pitfalls.
Important: the approach depends on having dense summary-level data with no significance thresholding (as is now commonly released with GWAS publications). We do not recommend running this out-of-the-box on data that has been pruned, thresholded, or restricted to top SNPs (consider single-marker tests for that). We recommend matching the GWAS SNPs to the SNPs in the LDREF/*.bim files as closely as possible, as only these SNPs will be used for prediction.
The functional reference data are loaded using the ./WEIGHTS/GTEx.Whole_Blood.pos which points to the individual *.RDat weight files, their gene identifiers, physical positions (and an optional PANEL column for the panel/study name). We have pre-computed *.pos files for all reference data downloadable below. Only weights in the file will be evaluated. The physical positions should correspond to the feature (e.g. TSS and TES) and will be used in the final output and for plotting.
Finally, we run FUSION.test.R using this data on chromosome 22:
Rscript FUSION.assoc_test.R \
--sumstats PGC2.SCZ.sumstats \
--weights ./WEIGHTS/GTEx.Whole_Blood.pos \
--weights_dir ./WEIGHTS/ \
--ref_ld_chr ./LDREF/1000G.EUR. \
--chr 22 \
--out PGC2.SCZ.22.dat
This should take under a minute and you will see weight names and physical positions printed to screen. If everything worked, this will generate the file PGC2.SCZ.22.dat with 73 rows, one of which is the header. We examine this file in detail in the following section.
Under the hood, the analysis steps are: (1) unify the GWAS and reference SNPs and remove/flip alleles as appropriate; (2) impute GWAS Z-scores for any reference SNPs that were missing using the IMPG algorithm; (3) estimate the functional-GWAS association statistic; (4) report results over all features tested.
Let’s look at the transcriptome-wide significant associations in PGC2.SCZ.22.dat (adjusted for 2058 genes in the GTEx whole blood reference) by calling cat PGC2.SCZ.22.dat | awk 'NR == 1 || $NF < 0.05/2058'. The first two lines are shown below, except I’ve transposed them to explain each entry:
| Column | Value | Usage | |
|---|---|---|---|
| 1 | FILE | … | Full path to the reference weight file used |
| 2 | ID | FAM109B | Feature/gene identifier, taken from --weights file |
| 3 | CHR | 22 | Chromosome |
| 4 | P0 | 42470255 | Gene start (from --weights) |
| 5 | P1 | 42475445 | Gene end (from --weights) |
| 6 | HSQ | 0.0447 | Heritability of the gene |
| 7 | BEST.GWAS.ID | rs1023500 | rsID of the most significant GWAS SNP in locus |
| 8 | BEST.GWAS.Z | -5.94 | Z-score of the most significant GWAS SNP in locus |
| 9 | EQTL.ID | rs5758566 | rsID of the best eQTL in the locus |
| 10 | EQTL.R2 | 0.058680 | cross-validation R2 of the best eQTL in the locus |
| 11 | EQTL.Z | -5.16 | Z-score of the best eQTL in the locus |
| 12 | EQTL.GWAS.Z | -5.0835 | GWAS Z-score for this eQTL |
| 13 | NSNP | 327 | Number of SNPs in the locus |
| 14 | MODEL | lasso | Best performing model |
| 15 | MODELCV.R2 | 0.058870 | cross-validation R2 of the best performing model |
| 16 | MODELCV.PV | 3.94e-06 | cross-validation P-value of the best performing model |
| 17 | TWAS.Z | 5.1100 | TWAS Z-score (our primary statistic of interest) |
| 18 | TWAS.P | 3.22e-07 | TWAS P-value |
Interpreting the output: This result indicates that the best performing prediction model for the gene was LASSO, which slightly outperformed the best eQTL (note a non-eQTL model is always used to compute the TWAS statistic even if the eQTL had higher accuracy). Over expression of the gene was positively associated with SCZ risk, which is consistent with the best eQTL SNP having a negative effect on expression and on GWAS. The TWAS Z-score was not more significant than the top GWAS SNP, which motivates conditional analyses to asses whether the locus harbors signal independent of the expression (see below).
The following tables list downloads for precomputed expression reference weights. Expression weights were typically computed from BLUP, BSLMM, LASSO, Elastic Net and top SNPs, except for instances where BLUP/BSLMM where excluded due to sample size or convergence issues. Each package contains a corresponding *.profile file listing performance statistics for each gene, as well as an *.err file summarizing performance and heritability across all genes.
| Tissue | Assay | # Samples | # Features | Study |
|---|---|---|---|---|
| Peripheral Blood | RNA array | 1,247 | 2,454 | [1] NTR |
| Whole blood | RNA array | 1,264 | 4,701 | [2] YFS |
| Adipose | RNA-seq | 563 | 4,671 | [3] METSIM |
| Brain (DLPFC) | RNA-seq | 452 | 5,420 | [4] CMC |
| Brain (DLPFC) | RNA-seq splicing | 452 | 7,772 | [4] CMC |
Each archive contains two sets of pos files, one for genes with significant heritability and one for all genes (labeled no_filter). Using genes that achieved significant heritability is recommended for typical analyses. Using weights from “All Samples” will also typically increase sensitivity, unless analyzing highly European-specific regions. A detailed comparison of models by population and GTEx version is provided here. Positions in the pos files are taken from GTEx annotations.
Weights were kindly estimated and provided by Junghyun Jung in the Mancuso lab.
| Tissue | All Samples | link | EUR Samples | link |
|---|---|---|---|---|
| Adipose - Subcutaneous | 581 | download | 479 | download |
| Adipose - Visceral (Omentum) | 469 | download | 393 | download |
| Adrenal Gland | 233 | download | 194 | download |
| Artery - Aorta | 387 | download | 329 | download |
| Artery - Coronary | 213 | download | 175 | download |
| Artery - Tibial | 584 | download | 476 | download |
| Brain - Amygdala | 129 | download | 119 | download |
| Brain - Anterior cingulate cortex (BA24) | 147 | download | 135 | download |
| Brain - Caudate (basal ganglia) | 194 | download | 172 | download |
| Brain - Cerebellar Hemisphere | 175 | download | 157 | download |
| Brain - Cerebellum | 209 | download | 188 | download |
| Brain - Cortex | 205 | download | 183 | download |
| Brain - Frontal Cortex (BA9) | 175 | download | 157 | download |
| Brain - Hippocampus | 165 | download | 150 | download |
| Brain - Hypothalamus | 170 | download | 156 | download |
| Brain - Nucleus accumbens (basal ganglia) | 202 | download | 181 | download |
| Brain - Putamen (basal ganglia) | 170 | download | 153 | download |
| Brain - Spinal cord (cervical c-1) | 126 | download | 115 | download |
| Brain - Substantia nigra | 114 | download | 100 | download |
| Breast - Mammary Tissue | 396 | download | 329 | download |
| Skin - Transformed fibroblasts | 483 | download | 403 | download |
| Blood - EBV-transformed lymphocytes | 147 | download | 113 | download |
| Colon - Sigmoid | 318 | download | 266 | download |
| Colon - Transverse | 368 | download | 294 | download |
| Esophagus - Gastroesophageal Junction | 330 | download | 275 | download |
| Esophagus - Mucosa | 497 | download | 411 | download |
| Esophagus - Muscularis | 465 | download | 385 | download |
| Heart - Atrial Appendage | 372 | download | 316 | download |
| Heart - Left Ventricle | 386 | download | 327 | download |
| Kidney - Cortex | 73 | download | 65 | download |
| Liver | 208 | download | 178 | download |
| Lung | 515 | download | 436 | download |
| Minor Salivary Gland | 144 | download | 114 | download |
| Muscle - Skeletal | 706 | download | 588 | download |
| Nerve - Tibial | 532 | download | 438 | download |
| Ovary | 167 | download | 138 | download |
| Pancreas | 305 | download | 243 | download |
| Pituitary | 237 | download | 219 | download |
| Prostate | 221 | download | 181 | download |
| Skin - Not Sun Exposed (Suprapubic) | 517 | download | 430 | download |
| Skin - Sun Exposed (Lower leg) | 605 | download | 508 | download |
| Small Intestine - Terminal Ileum | 174 | download | 141 | download |
| Spleen | 227 | download | 179 | download |
| Stomach | 324 | download | 260 | download |
| Testis | 322 | download | 272 | download |
| Thyroid | 574 | download | 482 | download |
| Uterus | 129 | download | 107 | download |
| Vagina | 141 | download | 120 | download |
| Whole Blood | 670 | download | 558 | download |
For reproducibility, legacy models from GTEx v7 are available for significant genes and all genes. Legacy models from GTEx v6 are also available for significant genes. See here for a comparison of GTEx v6 and v7 model performance.
Cross tissue weights for cross-tissue features generated through sparse canonical correlation analysis on GTEx gene expression version 6 and version 8.
| Study | # Features |
|---|---|
| GTEx v8 | 37920 |
| GTEx v6 | 37920 |
For more details and citation see: Feng, Helian, et al. “Leveraging expression from multiple tissues using sparse canonical correlation analysis (sCCA) and aggregate tests improves the power of transcriptome-wide association studies (TWAS).” 2021 PLOS Genetics.
Germline gene expression models constructed from tumor RNA-seq. See here for evaluation of per-cancer heritability.
| Cancer | # Samples | # Features |
|---|---|---|
| Manifest: significant | ||
| Bladder Urothelial Carcinoma | 380 | 1677 |
| Breast Invasive Carcinoma | 1049 | 4464 |
| Cervical Squamous Cell Carcinoma | 277 | 1117 |
| Colon Adenocarcinoma | 275 | 2098 |
| Esophageal Carcinoma | 170 | 697 |
| Glioblastoma Multiforme | 125 | 1039 |
| Head and Neck Squamous Cell Carcinoma | 499 | 2763 |
| Kidney Renal Clear Cell Carcinoma | 506 | 4139 |
| Kidney Renal Papillary Cell Carcinoma | 283 | 2080 |
| Brain Lower Grade Glioma | 483 | 4347 |
| Liver Hepatocellular Carcinoma | 352 | 1121 |
| Lung Adenocarcinoma | 500 | 2973 |
| Lung Squamous Cell Carcinoma | 464 | 2544 |
| Ovarian Serous Cystadenocarcinoma | 284 | 1620 |
| Pancreatic Adenocarcinoma | 172 | 1685 |
| Pheochromocytoma and Paraganglioma | 175 | 1342 |
| Prostate Adenocarcinoma | 468 | 4675 |
| Rectum Adenocarcinoma | 89 | 707 |
| Soft Tissue Sarcoma | 249 | 1091 |
| Skin Cutaneous Melanoma | 103 | 541 |
| Stomach Adenocarcinoma | 387 | 1718 |
| Testicular Germ Cell Tumors | 149 | 1296 |
| Thyroid Carcinoma | 484 | 5211 |
| Uterine Corpus Endometrial Carcinoma | 171 | 518 |
Expression and junction usage weights from Gusev, Lawrenson, et al. 2019 Nat Genet:
| Tissue & model | # Samples | # Features |
|---|---|---|
| Ovarian normal FTSEC expression | 60 | 542 |
| Ovarian normal OSEC expression | 105 | 608 |
| TCGA Ovarian tumor expression | 225 | 1745 |
| TCGA Ovarian tumor junction | 225 | 8760 |
| TCGA Breast normal expression | 90 | 1986 |
| TCGA Breast normal junction | 90 | 5392 |
| TCGA Breast tumor expression | 782 | 4466 |
| TCGA Breast tumor junction | 782 | 21569 |
| TCGA Prostate tumor expression | 457 | 4415 |
| TCGA Prostate tumor junction | 457 | 17858 |
Context specific weights computed using the CONTENT method for bulk expression across GTEx tissues and single-cell expression from California Lupus Epidemiology Study (CLUES). Each weight file contains a single model either CxC (context by context) or CONTENT (context specific) with the filename indicating the model subtype and cell/tissue type.
| Dataset | # Features |
|---|---|
| Manifest | |
| GTEx (CONTENT) | 278101 |
| GTEx (context-by-context) | 195607 |
| CLUES single-cell (CONTENT) | 9080 |
| CLUES single-cell (context-by-context) | 4314 |
For more details and citation see: Thompson et al. “Multi-context genetic modeling of transcriptional regulation resolves novel disease loci” pre-print
RWAS models for ATAC-seq activity from TCGA tumors (using raw data from Corces et al. 2018 Science). A description of the file structure is provided here. Each weight file contains models for lasso, lasso.as, lasso.combined, top1, top1.as, top1.combined, where the prefix lasso or top corresponds to penalized or top SNP models, and the suffix as or none or combined corresponds to allele-specific or total or combined activity models. Individual variant allele-specific results are also available here.
| Dataset | # Samples | # Features |
|---|---|---|
| TCGA pan-cancer RWAS models | 406 | 491037 |
For more details and citation see: Grishin et al. “Allelic imbalance of chromatin accessibility in cancer identifies candidate causal risk variants and their mechanisms” 2022 Nature Genetics (accepted)
FUSION format predictive weights computed by other groups/projects and generously made publicly available.
| Reference | Description | Link |
|---|---|---|
| Ratnapriya et al. 2019 Nat Genet | Retinal samples from 523 aged post-mortem human subjects from a spectrum of age-related macular degeneration (AMD) were RNA-seq profiled. | GEO |
| Gandal et al. 2018 Science | Postmortem human brain samples were collected as part of eight studies. | link |
The script for computing expression weights yourself works one gene at a time, taking as input a standard binary PLINK format file (bed/bim/fam) which contains only the desired SNPs in the cis-locus of the gene (or any other desired SNPs) and expression of the gene set as the phenotype. We typically restrict the cis-locus to 500kb on either side of the gene boundary.
A typical run looks like this:
Rscript FUSION.compute_weights.R \
--bfile $INP \
--tmp $TMP \
--out $OUT \
--models top1,blup,bslmm,lasso,enet
Assuming $TMP, $INP, and $OUT are defined, this will take as input the $INP.bed/bim/fam file and generate a $OUT.RDat file which contains the expression weights and performance statistics for this gene. Note that by default BSLMM is not enabled due to the intensive MCMC computation. We recommend running BSLMM on a sample of genes to evaluate performance and only retaining if accuracy is significantly higher than other models. For more information, see the /examples/GEUV.compute_weights.sh file in the FUSION package which details an entire batched run on publicly available data.
Under the hood, the detailed analysis steps are: (1) Estimate heritability of the feature and stop if not significant; (2) Perform cross-validation for each of the desired models; (3) Perform a final estimate of weights for each of the desired models and store results.
Important: Unless you plan to use your own LD-reference panel, we recommend first restricting your --bfile genotypes to the set of markers in the LD reference data we provide, as this is the set of SNPs that will be used for the final association statistic.
Optional: After all genes have been evaluated, make a WGTLIST file which lists paths to each of the *.RDat files that were generated and call Rscript utils/FUSION.profile_wgt.R <WGTLIST> to output a per-gene profile as well as an overall summary of the data and model performance. See the *.profile and *.err files bundled with any of the weights above for examples.
We often identify multiple associated features in a locus (or the same feature from multiple tissues) and would like to identify which are conditionally independent. Similarly, we may want to ask how much GWAS signal remains after the association of the functional is removed. The FUSION.post_process.R script performs post-processing on FUSION.assoc_test.R outputs to report these statistics. Continuing with the SCZ and GTEx whole blood example from above, let’s extract the transcriptome-wide significant associations and analyze them:
cat PGC2.SCZ.22.dat | awk 'NR == 1 || $NF < 0.05/2058' > PGC2.SCZ.22.top
Rscript FUSION.post_process.R \
--sumstats PGC2.SCZ.sumstats \
--input PGC2.SCZ.22.top \
--out PGC2.SCZ.22.top.analysis \
--ref_ld_chr ./LDREF/1000G.EUR. \
--chr 22 \
--plot --locus_win 100000
This will read in the expression weights for the selected genes, consolidate them into overlapping loci (using the boundary defined by --locus_win ), and generate multiple conditional outputs along with the following summary figure:
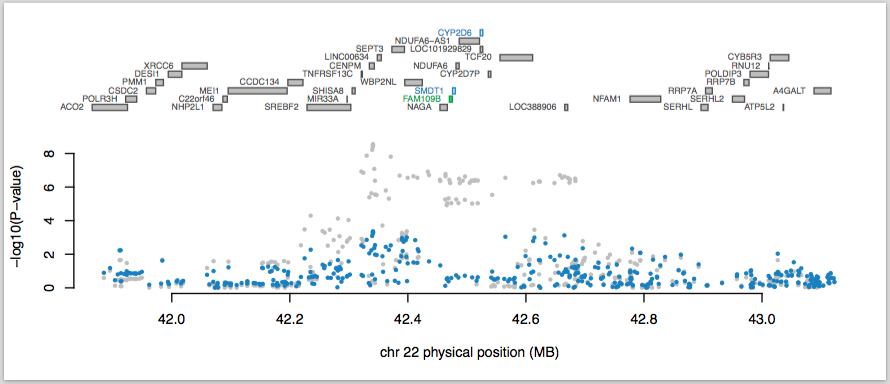
The top panel shows all of the genes in the locus. The marginally TWAS associated genes are highlighted in blue, and those that are jointly significant (in this case, FAM109B) highlighted in green. The statistics for the jointly significant genes are reported in the file PGC2.SCZ.TWAS.22.top.analysis.joint_included.dat (which contains the joint estimates and p-values) and the statistics for those genes that were dropped due to being conditionally non-significant are in the corresponding *.joint_dropped.dat file (which contains the conditional estimates and p-values).
The bottom panel shows a Manhattan plot of the GWAS data before (gray) and after (blue) conditioning on the green genes. You can see that this locus goes from being genome-wide significant to non-significant after conditioning on the predicted expression of FAM109B.
See additional FUSION.post_process.R --plot_* commands below. When working with many reference panels (such as GTEx), adding the --legend joint flag may be helpful to distinguish between panels.
Please see the FOCUS method and related paper of Mancuso et al. 2019 Nature Genetics for fine-mapping associated genes. FOCUS natively accepts results and weights from FUSION as well as PrediXcan.
Please see the MESC tool and corresponding pre-print of Yao et al. for a novel method to estimate the fraction of disease heritability that is mediated by all assayed/predicted gene expression.
The imputed association statistic is well-calibrated under the null of no GWAS association, but may be inflated from by-chance QTL co-localization when the GWAS locus is highly significant and LD is extensive. To assess this, we have proposed a permutation test (FUSION.assoc_test.R --perm <max runs>) which shuffles the QTL weights and recomputes an empirical association statistic conditional on the GWAS effects at the locus. This effectively tests if the same distribution of QTL effect sizes could yield a significant association by chance. The test is implemented “adaptively”, so permutation will stop after a sufficient number of significant observations (or at the maximum specified). This statistic is highly conservative (for example, truly causal genes can fail the test if their QTLs are in high LD with many other SNPs), and intended to prioritize associations that are already significant in the standard test for follow-up.
This adds the following columns to output: PERM.PV, the empirical permutation p-value (i.e. the fraction of permutations more significant than the observation); PERM.N, the total number of permutations performed, PERM.ANL_PV the analytical permutation p-value assuming permuted results are normally distributed, computing a Z-score to the observed results, and converting it to a two-tailed p-value.
You may be interested in computing a p-value for the joint association of a single gene/feature using predictors from multiple reference panels. Because the GWAS samples are always the same, this cannot be estimated using a simple meta-analysis. We have implemented a multiple degree-of-freedom omnibus test (FUSION.post_process.R --omnibus) which estimates and adjusts for the pairwise correlation between functional features. The flag looks for duplicate entries of the ID field in the --input file to select which predictors get combined into a single omnibus test.
In the case of N fully independent predictors this corresponds to an N-dof Chi^2 test. Because this is essentially a test for deviation of association statistics from the correlation matrix, care must be taken in evaluating biological plausibility by examining the pairwise correlations (also reported in a separate file).
An alternative to the TWAS test (which looks for a significant association between the predicted functional feature and the trait) is a colocalization test which looks for evidence of a shared causal variant between the functional feature and the trait. FUSION includes an interface to the COLOC software to compute an approximate colocalization statistic based on the marginal FUSION weights (this requires having the COLOC R package installed). The test is initiated with the Fusion.assoc_test.R --coloc_P flag, which sets the TWAS p-value threshold below which to compute COLOC statistics (we do not recommend computing this for non-significant TWAS associations).
Testing for colocalization requires specifying the functional (e.g. gene expression) study sample size to convert from association statistics back to effect-sizes, this can be done in two ways: (1) having an N column in the input file listing the sample size for each weight file; (2) Having a PANEL column in the input file and the --PANELN [FILE] flag pointing to a reference file with panel-specific sample sizes. The reference file must then contain PANEL and N columns listing the sample size for each expression panel. We recommend the second approach as the PANEL flag is useful in other analyses, but either approach yields the same computation. Finally, the --GWASN flag specifies the sample size of the GWAS study.
Effect sizes are then approximated for COLOC under the assumption that standard error is approximately inverse the square root of the sample size. PP0, PP1, PP2, PP3, PP4 columns will be added to the output which indicate the COLOC statistic for each of the COLOC hypotheses (no association, functional association only, GWAS association only, independent functional/GWAS associations, colocalized functional/GWAS associations).
When summary-based prediction is insufficient and individual-level data is available, FUSION can generate individual-level predictors using the utils/make_score.R script which are then directly usable in PLINK for prediction. This requires first running Rscript make_score.R [wgt.RDat file] > [SCORE_FILE] on the predictor weights file, and then running plink --bfile [GENOTYPES] --score [SCORE_FILE] 1 2 4 on the individual level data. As in the primary FUSION analysis, the score for the best perfomring model is used.
FUSION.compute_weights.R| Flag | Usage | Default |
|---|---|---|
| --bfile | Path to PLINK binary input file prefix (minus bed/bim/fam) | required |
| --out | Path to output files | required |
| --tmp | Path to temporary files | required |
| --covar | Path to quantitative covariates (PLINK format) | optional |
| --crossval | How many folds of cross-validation, 0 to skip | 5 |
| --hsq_p | Minimum heritability p-value for which to compute weights. | 0.01 |
| --hsq_set | Pre-computed heritability estimate (between 0.0-1.0), skips analysis. | optional |
| --models | Comma-separated list of prediction models | (see below) |
| --noclean | Do not remove temporary files (for debugging) | OFF |
| --PATH_gcta | Path to gcta executable |
gcta_nr_robust |
| --PATH_gemma | Path to gemma executable |
gemma |
| --PATH_plink | Path to plink executable |
plink |
| --pheno | Path to functional phenotype file (e.g. expression; PLINK format) | optional |
| --rn | Rank normalize the phenotype after QC (requires GenABEL and preprocessCore libraries) | OFF |
| --save_hsq | Save heritability results even if weights are not computed | OFF |
| --verbose | How much chatter to print to screen: 0=nothing; 1=minimal; 2=all | 0 |
| Gene expression models | |
|---|---|
| blup | Best Linear Unbiased Predictor computed from all SNPs. Requires GEMMA in path. |
| bslmm | Bayesian Sparse Linear Model (spike/slab MCMC). Requires GEMMA in path. |
| enet | Elastic-net regression (with mixing parameter of 0.5). |
| lasso | LASSO regression. Requires PLINK in path |
| top1 | Single best eQTL. |
FUSION.assoc_test.R| Flag | Usage | Default |
|---|---|---|
| --chr | Chromosome to analyze | required |
| --out | Path to output files | required |
| --ref_ld_chr | Prefix to reference LD files in binary PLINK format by chromosome | required |
| --sumstats | Path to LD-score format summary statistics (must have SNP, A1, A2, Z column headers) | required |
| --weights | File listing functional weight RDat files | required |
| --weights_dir | Path to directory where weight files | required |
| --coloc_P | P-value below which to compute COLOC statistic. Requires coloc R library. | OFF |
| --force_model | Force specific predictive model to be used. | OFF |
| --GWASN | Total GWAS/sumstats sample size for inference of standard effect size. | OFF |
| --max_impute | Fraction of GWAS SNPs allowed to be missing per gene (for IMPG imputation) | 0.5 |
| --min_r2pred | Minimum average IMPG imputation accuracy allowed | 0.7 |
| --PANELN | File listing PANEL and N for COLOC effect size computation | OFF |
| --perm | Maximum number of permutations to perform for each feature, 0=off | 0 |
| --perm_minp | If --perm, threshold to initiate permutation test | 0.05 |
FUSION.post_process.R| Flag | Usage | Default |
|---|---|---|
| --chr | Chromosome to analyze | required |
| --input | Path to TWAS test output | required |
| --out | Path to output files | required |
| --ref_ld_chr | Prefix to reference LD files in binary PLINK format by chromosome | required |
| --sumstats | Path to LDSC format summary statistics | required |
| --ldsc | Compute LD-scores across all features. This disables all other tests | OFF |
| --locus_win | How much to expand each feature (in bp) to define contiguous loci | 100,000 |
| --minp_input | Minium p-value to include feature in the analysis | 1.0 |
| --minp_joint | Minium p-value to include feature in the joint model | 0.05 |
| --max_r2 | Features with r^2 greater than this will be considered identical (and pruned) | 0.9 |
| --min_r2 | Features with r^2 less than this will be considered independent | 0.008 |
| --omnibus | Perform omnibus test for features with multiple models (disables other tests) | optional |
| --omnibus_corr | Only print pairwise correlations between models (model name or “best”) | optional |
| --plot | Generate pdf plots for each locus | OFF |
| --plot_corr | Plot correlation of genetic values for each locus (requires corrplot library) | OFF |
| --plot_eqtl | Plot marginal expression weight (approximately the eQTL beta) | OFF |
| --plot_individual | Plot conditional analyses for all individual predictors | OFF |
| --plot_legend | Add a legend to the plot for reference panels [options=all/joint] | OFF |
| --plot_scatter | Scatterplot of relationship between GWAS effect size and predictor | OFF |
| --report | Generate a report document with information on each locus (for twas-hub) | OFF |
| --save_loci | Save conditioned GWAS results for each locus | OFF |
| --verbose | How much chatter to print: 0=nothing; 1=minimal; 2=all | OFF |
What QC is performed internally on the GWAS summary data?
The methods automatically match up SNPs by rsID, remove strand ambiguous markers (A/C or G/T) and flip alleles to match strand to the weights and reference LD data. NO QC is performed on the functional/expression phenotype or the genotypes. We recommend the standard steps described in the GTEx documentation. For GWAS data, the IMPG algorithm is also used to infer statistics for any reference SNPs that do not have GWAS scores.
How careful do I have to be with samples/SNP matching across files?
As long as your sample and SNP identifiers are consistent across all the files, they will be matched and the intersection will be taken for all analyses. Order does not matter. This means the intersection of individuals is taken across all input files (bfile/pheno/covar) for the weights analysis. Column order does not matter for any file that has a header row.
Can I use my own LD reference panel?
Yes! The functional reference data is completely decoupled from the expression weights. If you are computing your own weights or have richer/unique LD data, simply convert it into by-chromosome binary PLINK files and point to them using the --ref_ld_chr flag. Standard caveats apply: the LD-reference should maximally cover the reference weight SNPs; should match the reference and GWAS populations; and increases in accuracy with sample size.
How can I validate the TWAS associations?
We recommend the same procedures for confirming TWAS associations as have been used in GWAS analyses: replication in an external study. This can be done at the level of individual associations, or in aggregate using a gene-based risk score (see manuscript for details).
We also proposed a permutation test in the manuscript the evaluates how significant the contribution from expression data is on top of the GWAS signal. Loci that pass this permutation test show evidence of heterogeneity that’s captured by the expression, and are less likely to be chance co-localization.
Lastly, the top eQTL in a gene is expected to explain a substantial fraction of the signal, so individual inspection of eQTLs and conditional analysis can help elucidate the underlying causal variant.
How should I interpret the effect direction?
The TWAS effect-size is an estimate of the genetic covariance between gene expression and the GWAS trait, so the direction of effect is informative of this relationship. If the phenotypes have not been treated unusually, then a positive effect means over-expression of the gene leads to an increase in the phenotype (and vice versa).
What if the GWAS statistics have highly variable sample sizes?
In principle having different sample sizes will violate the assumptions made in the IMPG/TWAS approaches and may lead to spurious results. Conservatively, we would recommend to restrict analyses to SNPs that have similar sample sizes, though we have not observed substantial differences in practice. If you do see differences, email us and we can work towards more specific recommendations.
How do I interpret warnings related to missing SNPs?
These warnings usually mean the GWAS summary statistics did not overlap with the expression weights to identify any SNPs with weights in common, or enough for the IMPG GWAS imputation to be performed to fill in missing statistics. We recommend double checking that your summary statistics overlap as closely as possible with the LDREF data. Predictions from genes with poor overlap will be unreliable.
What related software/data is available?
2022/02/01: Added GTEx v8 weights.
2018/04/08: Documentation for multiple post-processing flags and scripts (COLOC, plotting, individual-level prediction). Additional sanity checks for IMPG filling in of missing GWAS data.
2017/01/04: Added LD-score computation. METSIM weights were missing chr18.
2016/12/01: Formal FUSION release, complete codebase and reference overhaul.
We are grateful to the functional resources and all of the hard worked involved in study design, data acquisition, and analysis. Without the efforts of these groups our tools would be useless.
Data were generated as part of the CommonMind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffman-La Roche Ltd and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881 and R37MH057881S1, HHSN271201300031C, AG02219, AG05138 and MH06692. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories and the NIMH Human Brain Collection Core. CMC Leadership: Pamela Sklar, Joseph Buxbaum (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Keisuke Hirai, Hiroyoshi Toyoshiba (Takeda Pharmaceuticals Company Limited), Enrico Domenici, Laurent Essioux (F. Hoffman-La Roche Ltd), Lara Mangravite, Mette Peters (Sage Bionetworks), Thomas Lehner, Barbara Lipska (NIMH).
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health. Additional funds were provided by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Donors were enrolled at Biospecimen Source Sites funded by NCI\SAIC-Frederick, Inc. (SAIC-F) subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171), and Science Care, Inc. (X10S172). The Laboratory, Data Analysis, and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to The Broad Institute, Inc. Biorepository operations were funded through an SAIC-F subcontract to Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). The Brain Bank was supported by a supplements to University of Miami grants DA006227 & DA033684 and to contract N01MH000028. Statistical Methods development grants were made to the University of Geneva (MH090941 & MH101814), the University of Chicago (MH090951, MH090937, MH101820, MH101825), the University of North Carolina - Chapel Hill (MH090936 & MH101819), Harvard University (MH090948), Stanford University (MH101782), Washington University St Louis (MH101810), and the University of Pennsylvania (MH101822).
The Netherlands Study of Depression and Anxiety (NESDA) and the Netherlands Twin Register (NTR) were funded by the Netherlands Organization for Scientific Research (MagW/ZonMW grants 904-61-090, 985-10-002,904-61-193,480-04-004, 400-05-717, 912-100-20; Spinozapremie 56-464-14192; Geestkracht program grant 10-000-1002); the Center for Medical Systems Biology (CMSB2; NWO Genomics), Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL), VU University EMGO+ Institute for Health and Care Research and the Neuroscience Campus Amsterdam, NBIC/BioAssist/RK (2008.024); the European Science Foundation (EU/QLRT-2001-01254); the European Community’s Seventh Framework Program (FP7/2007-2013); ENGAGE (HEALTH-F4-2007-201413); and the European Research Council (ERC, 230374).
The Young Finns Study has been financially supported by the Academy of Finland: grants 286284 (T.L), 132704 (M.H.), 134309 (Eye), 126925, 121584, 124282, 129378 (Salve), 117787 (Gendi), and 41071 (Skidi); the Social Insurance Institution of Finland; Kuopio, Tampere and Turku University Hospital Medical Funds (grant X51001 for T.L.); Juho Vainio Foundation; Paavo Nurmi Foundation; Finnish Foundation of Cardiovascular Research (T.L.); Finnish Cultural Foundation; Tampere Tuberculosis Foundation (T.L., M.H.); Emil Aaltonen Foundation (T.L.); and Yrjö Jahnsson Foundation (T.L., M.H.)
The METSIM study has been supported by the Academy of Finland, the Finnish Diabetes Research Foundation, the Finnish Cardiovascular Research Foundation, the Strategic Research Funding from the University of Eastern Finland, Kuopio, Finland and EVO grant 5263 from the Kuopio University Hospital.
Logo by Ryan Beck from The Noun Project
 TWAS / FUSION
TWAS / FUSION